*This website was produced as an assignment for an undergraduate course at Davidson College.*
Review:
Creating Bacterial Strains from Genomes That
Have Been Cloned and Engineered in Yeast
Introduction:
Researchers at the J. Craig Venter organization described in a previous paper the "chemical synthesis, assembly, and cloning of a bacterial genome in yeast" (1). This information was accumulated to better understand the necessary steps to produce a synthetic cell. This synthetic cell production involves transforming a genome from yeast into a receptive environment that will execute the necessary functions. Lartigue's most recent paper attempts to complete their construction of a living microbe and essentially describes a method for performing genomic modifications outside of the host cell. The realm of possible genetic manipulations is far greater in yeast than in bacteria. Specifically, the Mycoplasma mycoides genome was cloned into a yeast plasmid that was then transplanted into Mycoplasma capricolum, a related species and convenient experimental organism, in order to generate a viable M. mycoides cell.
Figures:
Figure 1. Generation of Type III restriction enzyme deletions (1)
Panel A
(i) The first construct is the YCpMmyc1.1 clone, depicting the location of the typeIIIres gene and the two flanking genes. Via the fusion of two PCR products (CORE and TR), a linear DNA fragment, or knock-out cassette, was constructed. This cassette is shown below the YCpMmyc1.1 clone. CORE was comprised of a URA3 marker, SCEI endonuclease gene, and GAL1 promoter.
(ii) The knock-out cassette was then transformed into a yeast strain that carried the M. mycoides genome, in place of the Type III restriction enzyme gene (typIIIres) through a seamless deletion. The red bars above the typeIIImod gene and the tandem repeat sequence of the knock-out cassette indicate the positions that allowed for homologous recombination.
(iii) After the cassette was inserted into the genome, the researchers induced galactose, which, in turn, resulted in the expression of Sce I endonuclease. Sce I cleaved the site designated with the asterisk (*), and the deletion of this segment promoted homologous recombination. This ultimately resulted in the deletion of the knock-out cassette. These clones were then isolated by 5-fluoroorotic acid, a compound that selects against the URA3 gene.
Panel B
The arrows shown in panel A, upstream and downstream of the typeIIIres gene, indicate PCR primers (P299 and P302) that were used to verify the presence or absence of the cassette. Figure B is an agarose gel electrophoresis. Essentially, PCR was used to verify the changes made to the genome. The first lane is simply a kbp ladder. The gel shows the PCR products of the original genome (i), with cassette (ii), and without cassette (iii). Lanes 2-4 represent the constructs “in yeast.” These lanes serve as positive controls, demonstrating what happens to the YCpMmyc1.1 genome in yeast. Lanes 5-8 represent the constructs “post-transplant,” or after having been transplanted into M. capricolum. The results are expected. The band lengths are fairly consistent between the constructs “in yeast” and “post-transplant.” The bands of the constructs with the knock-out cassette display a longer kbp length than the original clone (didn't travel as far on the gel), while the bands of the constructs without the knockout cassette demonstrate a shorter kbp length than the original clone (traveled farther).
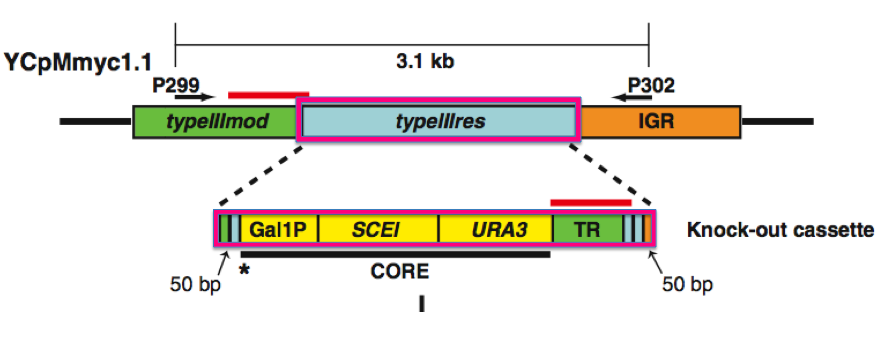
Figure 1. The knock-out cassette (bottom magenta box) is the construct that was inserted into the genome in place of the typeIIIres gene (upper magenta box).
Image from Lartigue et al., 2009.
Table 1. Transplantation of M. mycoides YCp genomes from yeast into wild-type and RE(-) M. capricolum recipient cells (1)
This table demonstrates the successes and failures of transplantation. The cloned genome was isolated from yeast and transplanted into wild-type M. capricolum cells. No transplants were recovered, which the researchers hypothesized may have been due to a restriction endonuclease in the recipient cells. After inactivating the restriction enzyme in M. capricolum and protecting the donor DNA by in vitro methylation, the cloned genome was once again isolated from yeast and transplanted into both wild-type and RE(-) M. capricolum. Transplantations were successful in both recipient cells. The yeast strain, genome, methylation treatment, and number of blue, tetracycline-resistant colonies counted after transplantation are indicated in the table. When M. capricolum was used as the recipient for YCpMmyc1.1, colonies were obtained when the donor DNA was untreated, treated with M.capricolum extracts, treated with M. mycoides extracts, mock-methylated, and treated with purified methylases. The transplantation of YCpMmyc1.1 into wild-type M. capricolum cells resulted in no colonies when the donor DNA was mock-methylated or untreated. The W303a yeast strain was transplanted during the various stages of the knockout process. All transplants into M. capricolum RE(-) were successful, except for the genome in which 500 bp of YCpMmyc1.1 were deleted; this was expected because this served as the negative control.
Figure 2: Southern blot analysis of M. mycoides transplants (1)
Panel A- The transplant colonies were probed with a M. mycoides-specific element (IS1296). Southern blot analysis was used to verify that the recovered colonies were M. mycoides cells and that the genome was conserved through transplantation.
- Lane 1 serves as a negative control because wild-type M. capricolum is probed with a M. mycoides specific probe. No bands are expected, and no bands are present.
- Lane 2 serves as a positive control because the native M. mycoides with YCpMmyc1.1 is being probed with a M. mycoides-specific element. Bands are expected, and bands are present.
- Lane 3 shows that M. mycoides, transplanted with YCpMmyc1.1, binds to the probe. In addition, the similar banding patterns between lanes 2 and 3 demonstrate that the transplanted genome was not altered.
- Lane 4 shows the probe binding to the construct with the knockout cassette in place of the typeIIIres gene. The bands appear similar to those found in lanes 2 and 3.
- Lane 5 shows the probe binding to the construct with the deletion of the typeIIIres gene. Although there is slight shifting, the banding pattern is similar to those in lanes 2, 3, and 4.
Panel B- The engineered transplants were then tested via Southern blot analysis to verify that the Type III restriction enzyme gene had been deleted. The transplants were probed with the typeIIIres gene sequence; this analysis verified that the gene was absent in the engineered transplants but present in the native M. mycoides genome.
- Lane 1 serves as the negative control. No band is observed because the typeIIIres probe does not bind to wild-type M. capricolum.
- Lane 2 serves as the positive control. A band is observed where the typeIIIres probe binds to the native M. mycoides cell with YCpMmyc1.1.
- Lane 3 verifies that the gene is absent in the engineered transplants. No band is observed for the construct with the knockout cassette in place of the typeIIIres gene.
- Lane 4 shows no band and demonstrates that the typeIIIres gene has been deleted.
Panel C- The sequence of the typeIIIres deletion confirmed that the gene was removed as designed. The green portion of the sequence is the typeIIImod gene, the blue portion is the remaining typeIIIres gene, and the orange portion is the intergenic region. The red boxes designate start and stop codons of the typeIIIres gene, and the black box designates the stop codon for the typeIIImod gene.
Figure 2. This Southern blot was used to verify that the Type III restriction enzyme gene had been deleted. In order to confirm that the gene had been deleted, the constructs were probed with the typeIIIres gene sequence. Lane two served as the positive control, and the band (labeled by the red box) is the only band that was expected to be observed.
Image from Lartigue et al., 2009.
Figure 3: Moving a bacterial genome into yeast, engineering it, and installing it back into a bacterium by genome transplantation (1)
This figure depicts the method of transferring a genome between species. Specifically the circular diagram demonstrates that a yeast vector is transformed into a bacterial genome, the bacterial genome is isolated and transplanted into a eukaryotic (yeast) cell where it is manipulated, and the engineered genome is transplanted back into bacteria. This process can be performed multiple times to create many possible modifications, including insertions, deletions, and rearrangments.
Critique:
I thought this paper provided a very thorough description of the steps taken in their work. The researchers provide a very convincing argument and significant evidence for their case. I found the schematic of the various constructs in Figure 1A to be particularly helpful, as they provided the main foundation of the study and were constantly referred to throughout the paper. Table 1 was compact and easy to follow, and I thought it was informative that the researchers included their initial failure during transplantation from yeast to wild-type M. capricolum cells. Both their initial failure and the description of how they overcame the obstacle were insightful for those pursuing future research. Figure 2A and 2B were very well controlled in themselves and offered control to the experiment as a whole. Figure 3, while not compelling evidence for their argument, presented a nice schematic for the “big picture.”
I found it confusing that the cloned genome, YCpMmyc1.1, is comprised of a long series of confusing letters and numbers. In addition, the genome of this clone is referred to by this title regardless of the cellular source. Thus, YCpMmyc1.1 can refer to the genome in the original M. mycoides strain, the genome in yeast, the genome transplanted from M. mycoides or yeast, or the genome as DNA from any of the sources above. Perhaps better distinguishing references would eliminate confusion for the reader.
Figure 1B is one figure that could use a few improvements. First, the varying brightness of the bands is problematic. A simple loading control would be valuable. In addition, while this figure employs a good positive control, there is no negative control. It would have been beneficial to have a lane of PCR with DNA that is not specific for the primers. This would ensure that there is no nonspecific binding occurring.
Table 1 contains a great deal of information and is very well organized. However, the main piece of information that the reader is supposed to gather is the number of tetracycline-resistant, blue colonies obtained after transplantation. Perhaps an actual demonstration of the colonies, or lack of colonies, would have been more clear and captivating for the reader. In addition, I would have liked to see some explanation of the diverse range of numbers of transplants or colonies. The number of colonies observed ranged from 9±4 for the VL6-48N yeast strain transplanted with YCpMmyc1.1 (methylated with M. capricolum extracts) to 52±12 for the W303a yeast strain, transplanted with untreated YCpMmyc1.1-typeIIIres. An explanation for this wide range of values would be informative.
Future Work:
This study has laid the groundwork for isolating, manipulating, and transforming DNA among species. One of the most obvious, but ambitious, implications of these findings would be in gene therapy, a technique used for correcting defective genes that often result in disease. Currently, there are several approaches, but this new method appears, in theory, to be a likely approach as well. It would be logical to move the organism’s genome into yeast, manipulate the DNA by replacing the abnormal gene with a normal gene, and transplant the genome back into the organism. There are many steps to be taken between this study and ultimately using this method for gene therapy. A few areas to begin exploring are described below.
One area to explore is the possibility of creating various manipulations and engineering new modifications to the genome. These modifications could include insertions, deletions, and rearrangements. Incorporating the same methods used in this paper, one could begin to test the limit of manipulating the DNA. For example, to replace a maladaptive gene with a “normal” gene, I would use the same methods described in this paper. I would use homologous recombination to replace the maladaptive gene with the “normal” gene of interest. I would use PCR and gel electrophoresis to verify the presence or absence of the "normal" gene of interest. To ensure that the genome was conserved during transplanation, I would then use a Southern blot, probed with a sequence that binds to the “normal” gene of interest. Up to this point, the methods described in the paper would be employed (see above). One addition to the study would be a Western blot. This functional would be used to verify that protein production, with the “normal” gene of interest having been incorporated, is actually taking place. This step is critical because the gene may have inserted and been transplanted, but it is useless if it is not functional. If no band appeared on the Western blot, indicating no protein production, steps would be taken to determine why the protein is not functional and what can be done to fix the problem.
Another area that would require exploration is the feasibility of experimental organisms. Experimenting with other strains of yeast and bacteria would be beneficial in gaining an understanding of what may be possible for this new method. The transplantation obstacle in this paper gives insight into the fact that there may be species-specific hurdles to face when transferring a genome between “branches of life” (1). One might start by performing the same research done in this paper but using different organisms, such as E. coli.
Lastly, transforming the manipulated genome back into the original host would be beneficial. In this study, the genome from M. mycoides was cloned, manipulated, and transformed into a different organism, M. capricolum. While the paper mentioned that M. capricolum was convenient for the recipient cell due totheir fast growth rate, it would be worth the time and effort to attempt to transplant the manipulated genome back into the original host. These three fundamental subjects of research can use many of the same methods and tests used this paper, with a few minor experimental changes. These studies will likely lead to a better understanding of the limits of this method in the areas of “pathogenicity and biology of mycoplasmas” (1). A better understanding of mycoplasmas could “accelerate the construction of live vaccine strains," and in the long run, this method may contribute to an advance in gene therapy (1).
References:
(1) Lartigue C, Vashee S, Algire M, Chuang R, Benders G, Ma L, Noskov V, Denisova E, Gibson D, Assad-Garcia N, Alperovich N, Thomas D, Merryman C, Hutchinson III C, Smith H, Venter J, Glass J. 2009. Creating bacterial strains from genomes that have been cloned and engineered in yeast. Science 325: 1693-1696.
Davidson College Molecular Biology Homepage
Please send any questions, comments, or suggestions to moheinzelmann@davidson.edu.